Enzymologie pathologique
| MALADIE DE GAUCHER |
Sphingolipidose secondaire à un déficit en glucosylcéramidase. Entraîne une accumulation de glucosylcéramide dans les cellules du système réticulo-endothélial. La plus fréquente des maladies génétiques lysosomales.
Epidemiologie :Affection héréditaire autosomique récessive, 30 fois plus fréquente chez les juifs ashkénazes 1/2500 naissances dans ce groupe ethnique .
Signes clinques:
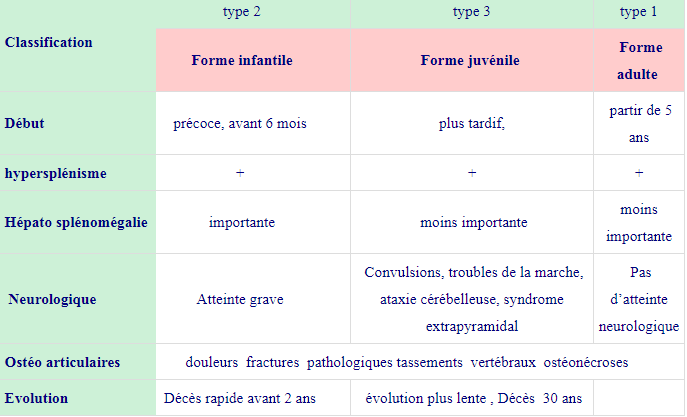
| Classification | type 2 | type 3 | type 1 |
| Forme infantile | Forme juvénile | Forme adulte | |
| Début | précoce, avant 6 mois | plus tardif, | partir de 5 ans |
| hypersplénisme | + | + | + |
| Hépato splénomégalie | importante | moins importante | moins importante |
| Neurologique | Atteinte grave | Convulsions, troubles de la marche, ataxie cérébelleuse, syndrome extrapyramidal | Pas d’atteinte neurologique |
| Ostéo articulaires | douleurs fractures pathologiques tassements vertébraux ostéonécroses | ||
| Evolution | Décès rapide avant 2 ans | évolution plus lente , Décès 30 ans | |
| Diagnostic: • Myélogramme: mis en évidence des cellules de Gaucher . • Biopsie hépatique : mis en évidence des cellules de Gaucher . • Mise en évidence du déficit enzymatique (leucocytes, fibroblastes) • Elévation des phosphatases acides Traitement: • Splénectomie parfois nécessaire • Une enzyme de substitution a été récemment mise au point: glucocérébrosidase (IV). |