Sclérose latérale amyotrophique
En 1874, Charcot décrivit complètement cette maladie nouvelle, description basée sur une vingtaine de cas cliniques et cinq autopsies recueillis à l’hôpital de la Salpêtrière. cette affection porte désormais le nom de sclérose latérale amyotrophique.
Définitions : Sclérose latérale amyotrophique SLA ou maladie de Charcot est une affection dégénérative du motoneurone ou encore du système moteur font momentanément partie d’une même entité. ( maladies du motoneurone ou motor neuron diseases ).
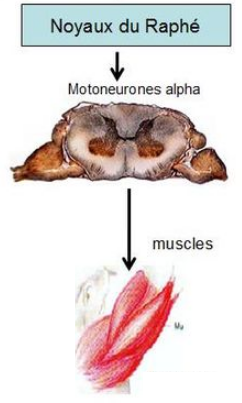
La SLA est due à une dégénérescence progressive de certaines cellules nerveuses (ou neurones), les motoneurones. Les motoneurones permettent de transmettre les ordres de mouvement (envoyés par le cerveau) aux muscles qui vont effectuer le mouvement commandé (figure 1). Ces neurones sont situés dans le cerveau (dans la zone appelée cortex moteur), dans la moelle épinière, qui est une sorte de cordon situé à l’intérieur de la colonne vertébrale, prolongeant le cerveau, ainsi que dans le bulbe rachidien (qui assure la jonction entre le cerveau et la moelle épinière).
Classifications : ce groupe comprend
- la SLA,
- forme sporadique et familiale
- la paralysie bulbaire progressive (PBP)
- l’atrophie musculaire progressive (AMP)
- la sclérose latérale primitive (SLP)
- la forme juvénile de SLA
- l’amyotrophie monomélique
C’est en 1990, en Espagne, à El Escorial, que s’est tenue la réunion de la Fédération mondiale de neurologie pour mettre au point ces critères.
Épidémiologie :
Facteurs de risque :
Le facteur racial est diversement apprécié.
Dans les études faites aux États-Unis, la résistance de la population noire a été remarquée.
Quant aux régions à haute incidence de SLA dans le Pacifique Ouest, le facteur racial ne semble pas être déterminant.
D’autres facteurs de risque ont été rapportés avec une fréquence variable.
Dans les antécédents des patients atteints de SLA, la fréquence de traumatismes répétés a été soulignée par de nombreux auteurs. Mais dans l’étude de Chancellor et al, les traumatismes non accompagnés de fracture ne seraient pas associés à une plus grande fréquence de SLA.
En revanche, Gresham et al, dans une étude cas-contrôles, n’ont pas observé d’association entre fractures et incidence de la SLA.
Parmi les autres facteurs, il faut citer les activités physiques intensives, la vie en communauté rurale.
Ces deux dernières études ont aussi montré l’association significative entre l’exposition au plomb et le développement de la SLA.
Dans une étude rétrospective portant sur 560 cas de maladies du motoneurone, Li et al n’ont trouvé aucune association significative entre divers paramètres, notamment traumatismes, fractures, expositions variées et maladie du motoneurone, à l’exception d’une tendance à une moindre fréquence pour les patients ne consommant pas d’alcool.
Enfin, il faut signaler la possible association entre SLA et antécédents de chocs électriques.
Des facteurs environnementaux inconnus sont peut-être à l’origine des cas conjugaux observés et des observations de survenue quasi simultanée de SLA chez des sujets sans liens familiaux, et cela dans un même lieu géographique, ont été rapportées.
Enfin, il faut signaler les récentes études sur la plus grande fréquence de la SLA chez les sujets en contact avec des animaux domestiques, en particulier les chiens.
A – Incidence :
L’incidence est le nombre de nouveaux cas survenant chaque année pour 100 000 habitants.
Pour la SLA, elle varie de 0,96 à 2,25. La moyenne se situerait autour de 1,5.
Généralement, le taux d’incidence augmente dans tous les pays avec l’âge jusqu’à un pic situé entre 60 et 75 ans, suivi d’un bref déclin après.
L’incidence paraît identique dans les populations noires et blanches.
B – Répartition selon le sexe et prévalence :
Le rapport hommes/femmes (H/F) varie selon les pays et les publications .
Il est en moyenne de 1,5/1. Cette prépondérance masculine pourrait résulter d’un facteur hormonal ou de l’effet cumulatif d’autres facteurs tels les traumatismes, l’activité physique ou des facteurs dus à l’exposition professionnelle.
La prévalence est définie comme le nombre de cas de la maladie observés à un moment donné, habituellement pour 100 000 habitants.
La moyenne de la prévalence de la SLA pour les pays occidentaux se situerait autour de 4 à 6 pour 100 000 habitants.
C – Mortalité :
Les taux de mortalité pour la SLA, ou maladie du motoneurone, sont obtenus par examen des causes de décès publiées dans la plupart des pays.
Le taux de mortalité varie de 0,5 à 1,1 pour 100 000 habitants, centré autour de 0,8 avec une homogénéité remarquable dans tous les pays.
L’ augmentation de la mortalité a pu être expliquée par différents facteurs : une meilleure connaissance de la maladie de la part des médecins, une augmentation du nombre de neurologues, l’effet Gompertzien de compétition intermaladies et une survie prolongée due aux meilleurs traitements des affections comme les accidents vasculaires ou cardiaques, permettant l’expression de la maladie SLA chez les patients âgés survivants.
Aspects cliniques :
A – Âge de début :
L’âge moyen de début se situe autour de 60 ans, avec des variations mineures dans les différentes séries.
L’âge de début diffère sensiblement selon le type de la maladie.
Il est sensiblement plus élevé dans les formes à début bulbaire et chez les femmes, et plus bas dans les formes à type d’atrophie musculaire progressive.
Les formes à début précoce ne sont pas rares, elles sont néanmoins rencontrées plus volontiers dans les formes familiales.
B – Durée d’évolution, pronostic et facteurs prédictifs de l’évolution :
La durée moyenne d’évolution de la SLA varie entre 2 et 3 ans.
La durée d’évolution semble liée à la forme clinique de la maladie du motoneurone, aux signes initiaux, à l’âge de début des troubles, au délai séparant le début des troubles et le diagnostic.
Dans les études récentes, l’effet de l’âge de début montre une nette différence : pour un début situé entre 25 et 44 ans, la survie médiane est de 71,5 mois, alors que pour un début entre 45 et 54 ans, il est de 35 mois et enfin entre 55 et 74 ans, il est de 32,5 mois.
Cette différence de durée de survie en fonction de l’âge de début n’a pas été confirmée par toutes les études.
Dans l’étude de Norris et al, la survie dépasse 100 mois dans 40 % des formes à début aux membres supérieurs, 50,1 % pour les formes à début aux membres inférieurs et seulement 6,2 % pour les formes à début bulbaire.
La survie à 5 ans se situe entre 10 et 30 % des cas.
Elle est très nettement plus basse dans les formes à début bulbaire (aucun cas dans la série de Chancellor et al) ou dans les formes spinales avec atteinte bulbaire développée au cours de l’évolution (11 % des cas, pour une valeur de 28 % pour la totalité de la population étudiée).
D’autres séries n’ont pas confirmé la relation entre durée d’évolution et forme à début bulbaire ou spinal.
Le délai entre le début apparent des troubles et le diagnostic apparaît aussi comme un facteur pronostique majeur dans la série de Lee et al.
Finalement, en comparaison avec les études plus anciennes, la durée de survie de la SLA ne s’est pas sensiblement modifiée : Mulder et Howard reportaient une survie à 3 ans dans 50 %des cas, à 5 ans dans 20 % des cas et à 10 ans dans 10 % des cas.
D’autres facteurs prédictifs ont été analysés, se basant sur des systèmes de scores utilisant différentes variables comme l’âge de début, l’âge au diagnostic, la forme de début, le score clinique utilisé, les fonctions pulmonaires et d’autres variables biologiques ou électrophysiologiques.
Ainsi, le degré de détérioration clinique et pulmonaire serait un test fiable pour prédire la durée de survie.
Le degré de perte motoneuronale a été montré comme linéaire et symétrique au cours de l’évolution de la SLA, non influencé par l’âge de début ou le site de début.
Ce qui permit à Jablecki et al de prédire la durée de survie approximative pour un individu donné selon l’âge de début, la durée d’évolution du déficit moteur et l’estimation de son état clinique mesuré par un système de score approprié.
D – Étude clinique :
1- Forme commune : le début brachial est le plus fréquent.
2- Forme à début brachial :
* Déficit musculaire :
Les signes et symptômes initiaux de la maladie sont localisés aux membres supérieurs dans 30 à 40 %des cas.
Le déficit musculaire débute à l’extrémité distale d’un membre dans la plupart des cas.
Le début proximal est plus rare (environ 10 % des cas).
Les troubles progressent ensuite et suivent une évolution ascendante.
Les mouvements volontaires des doigts et notamment du pouce sont les premiers touchés.
Avant que ne se développe l’atrophie musculaire, le patient se plaint fréquemment d’une maladresse dans les gestes les plus courants ; parfois, c’est une raideur de la main ou encore des phénomènes crampiformes, volontiers déclenchés par le froid.
L’atrophie des muscles intrinsèques de la main est donc très souvent le premier signe à apparaître.
L’amyotrophie siège préférentiellement au court abducteur du pouce et aux interosseux.
Les espaces métacarpiens se creusent, particulièrement le premier espace.
Progressivement, l’atrophie gagne tous les muscles de la main, le pouce se met sur le même plan que les autres doigts, réalisant la « main de singe ».
La main peut parfois se déformer en griffe avec extension de la première phalange et flexion des autres.
L’atrophie gagne ensuite les muscles de l’avant-bras, touchant d’abord les fléchisseurs des doigts puis les extenseurs.Au bras, le biceps est atteint avant le triceps.
Enfin, elle atteint les muscles de la ceinture scapulaire.
La marche de la maladie n’est pas toujours régulièrement ascendante et les déficits amyotrophiques peuvent sauter, par exemple, de la main à l’épaule.
L’atrophie atteint également les muscles du tronc et de la nuque.
Dans la majorité des cas, l’atteinte est franchement asymétrique au début et peut en imposer pour une pathologie nerveuse locale (paralysie cubitale ou médiane).
Le début par une atteinte pseudoradiale est beaucoup plus rare mais souvent trompeur.
L’atrophie gagne le membre supérieur opposé tandis que progresse l’atteinte du membre initialement atteint.
Assez rapidement, les deux membres supérieurs sont paralysés et dans certains cas, aucun mouvement des membres supérieurs n’est possible.
Les membres sont décharnés, l’aspect du malade est impressionnant, d’autant qu’il s’associe fréquemment un amaigrissement marqué pas toujours expliqué par la fonte musculaire ou les troubles de déglutition.
Cette perte du tissu graisseux avait été notée comme caractéristique de l’affection par les anciens auteurs.
* Crampes :
Elles précèdent souvent le déficit ou l’atrophie musculaire ; elles sont diversement appréciées selon les auteurs.
Fréquentes dans la plupart des séries, jusqu’à 72 % des cas, elles sont rares dans d’autres.
Elles peuvent précéder l’apparition de l’amyotrophie de plusieurs mois, puis tendent à disparaître lorsque progresse l’amyotrophie.
* Fasciculations :
Elles sont un des signes précoces et caractéristiques de la maladie.
Les premiers auteurs les avaient déjà notées et les nommaient fibrillations.
Ces soubresauts musculaires correspondent à des décharges asynchrones et le plus souvent arythmiques des fibres nerveuses motrices.
Leur origine n’est pas totalement élucidée.
Elles proviendraient d’une hyperactivité motoneuronale siégeant dans le corps cellulaire ou plus probablement au niveau axonal.
Elles constituent donc un signe précoce de la maladie et peuvent inaugurer celle-ci dans certains cas : 3,5 % des cas pour Gubbay et al.
Leur présence au niveau de muscles encore sains et surtout dans des territoires éloignés ou inhabituels, tels le thorax ou la langue, est très évocatrice de la maladie.
Les fasciculations sont généralement limitées à une fibre musculaire ou plus souvent à un groupe de fibres appartenant à la même unité motrice.
Le soubresaut musculaire est ainsi assez limité et discret, moins grossier que dans les myokymies.
Elles peuvent cependant être plus larges, confluer et entraîner ainsi des mouvements ondulatoires musculaires très nettement visibles.
À l’extrême, elles sont généralisées et peuvent en imposer pour une chorée fibrillaire de Morvan.
Le plus souvent, au début, elles sont limitées à certains muscles tel le premier interosseux dorsal.
Elles sont indolores et pas toujours perçues par le patient.
Parfois, leur abondance et leur intensité entraînent des mouvements des segments de membre.
Elles persistent pendant le sommeil.
L’exposition au froid ou la percussion directe du muscle sont des méthodes classiques pour les rechercher.
Elles sont présentes à un moment ou un autre de la maladie dans près de 90 % des cas.
Elles ont tendance à diminuer puis disparaître lorsque progresse l’amyotrophie.
* Existence d’un syndrome pyramidal :
Il vient ajouter un élément très particulier à cette amyotrophie progressive.
Il lui confère sa singularité clinique et sa présence confirme, s’il en était besoin, le diagnostic de SLA.
En effet, il est surprenant de constater, au niveau des territoires amyotrophiés (évoquant une atteinte du système nerveux périphérique), une hyperréflectivité tendineuse ou même la simple conservation de réflexes qui devraient être normalement abolis.
Dans la forme commune de la maladie, le syndrome pyramidal est peu prononcé au début.
On note cependant l’existence de réflexes vifs, polycinétiques et diffusés, le plus souvent seulement aux membres inférieurs alors qu’ils sont simplement conservés aux membres supérieurs.
Dans certains cas, ils sont diminués ou même abolis.
L’hypertonie pyramidale est rarement marquée aux membres supérieurs, elle est le plus souvent masquée par l’amyotrophie.
Elle peut être plus nette aux membres inférieurs et entraîner une démarche spastique.
Le clonus de la rotule et la trépidation épileptoïde du pied peuvent alors être notés.
Mais le signe de Babinski est inconstant, son absence en présence d’un syndrome pyramidal patent serait un élément diagnostique important.
La progression de l’atrophie musculaire aux membres inférieurs et sa localisation aux muscles distaux des jambes tend à faire disparaître le signe de Babinski, dans 17 % des cas dans l’étude de Meininger et al.
En effet, le caractère progressif de cette affection entraîne une extension de l’atrophie musculaire qui gagne ainsi les membres inférieurs et surtout les muscles d’innervation bulbaire.
* Atteinte bulbaire :
Elle est effectivement pratiquement constante au cours de la SLA.
Elle en commande le pronostic.
C’est en effet le plus souvent des conséquences de l’atteinte bulbaire que meurent les malades.
En fait, il s’agit d’une atteinte bulbaire directe due à la dégénérescence des noyaux bulbaires et d’une paralysie pseudobulbaire due au syndrome pyramidal.
C’est le mélange de ces deux syndromes qui confère à l’affection ses caractères propres.
L’atteinte bulbaire est parfois présente dès le début des troubles atrophiques des membres supérieurs et l’examen de la langue doit être systématiquement pratiqué afin de découvrir les fasciculations linguales qui apportent alors la quasi-certitude du diagnostic.
Le patient peut ne présenter au début qu’une gêne de la parole ou de discrets troubles de la déglutition.
Mais les troubles bulbaires progressent rapidement.
La voix devient mal articulée, nasonnée et finalement incompréhensible, réduite à quelques sons.
Ce trouble de la parole est dû à la fois à des phénomènes dysarthriques et à des troubles dysphoniques.
En effet, non seulement le trouble phonatoire est dû à l’atteinte de la langue, du voile du palais et des muscles péribuccaux, mais aussi à un trouble phonatoire résultant d’une faiblesse expiratoire.
La langue s’atrophie, d’abord latéralement puis en masse.
Elle est parcourue d’incessantes fasciculations et sa mobilité devient nulle.
Le voile du palais est flasque ou peu mobile, mais le réflexe du voile ainsi que le réflexe nauséeux persistent longtemps.
Les muscles de la face sont également touchés : orbiculaires des lèvres et houppe du menton.
Les masséters sont atteints et s’atrophient, mais la mastication est longtemps conservée.
Cette localisation des troubles donne au patient un aspect très particulier : visage émacié, quasi cadavérique, où seuls les muscles oculomoteurs fonctionnent.
Les troubles de la déglutition ajoutent à ce tableau dramatique d’un patient aphémique les risques de fausses routes et de dénutrition.
Au début, la déglutition est lente et pénible, le malade s’engoue, les liquides refluent par le nez, les fausses routes deviennent plus fréquentes rendant l’alimentation longue et pénible, contribuant ainsi à la cachexie si souvent rencontrée dans cette affection.
La gêne respiratoire est plus souvent discrète mais peut être au premier plan dans certains cas.
L’atteinte périphérique bulbaire pure est donc caractérisée par l’atrophie musculaire linguale et les fasciculations et le syndrome pseudobulbaire par la vivacité du réflexe nasopalpébral et surtout du réflexe massétérin.
Le rire et pleurer spasmodique typique des syndromes pseudobulbaires est parfois présent.
Il est enfin possible que le syndrome pseudobulbaire inaugure l’atteinte bulbaire par des troubles phonatoires et de déglutition sans que l’examen puisse découvrir de fasciculations ou d’atrophie musculaire.
* Évolution :
L’évolution de cette forme à début brachial ne dépasse guère 2 ou 3 ans, bien qu’existent des cas à évolution prolongée.
Le tableau typique de la SLA dans sa forme commune comprend ainsi une tétrade caractéristique : atrophie musculaire progressive, syndrome pyramidal, fasciculations et, à un moindre degré, crampes.
Cette tétrade que l’on pourrait appeler positive est complétée par une tétrade négative : absence de troubles sensitifs, de troubles oculomoteurs, de troubles sphinctériens et d’escarres.
Ces signes négatifs sont d’une grande importance pour le diagnostic, mais ils peuvent manquer, nous le verrons aussi plus loin.
3- Modalités de début :
Si le début brachial est le plus fréquent, d’autres modalités de début sont possibles : l’atrophie peut débuter aux membres inférieurs.
L’atteinte est également asymétrique, siégeant soit à la racine, soit à l’extrémité, pouvant réaliser dans ces cas de véritables formes pseudopolynévritiques.
Dans certains cas, l’atteinte initiale se situe au niveau du tronc et du cou.
Ailleurs, l’atteinte est qualifiée de diffuse d’emblée. L’atteinte bulbaire initiale est fréquente. Elle pose au début des problèmes diagnostiques avec d’autres affections telle la myasthénie.
Mais rapidement, le tableau s’enrichit suffisamment pour rendre ce diagnostic aisé.
Le début bulbaire est retrouvé dans près de 30 % des cas.
Ces modalités de début, la présence ou l’absence d’un des signes essentiels de la maladie font autant de formes cliniques dont la diversité ne doit pas nous faire oublier que le tableau clinique de maladie du motoneurone est suffisamment caractéristique pour ne pas laisser place au doute.
E – Formes cliniques :
1- Sclérose latérale amyotrophique à forme pseudopolynévritique :
Le début s’annonce par des phénomènes subjectifs à type d’engourdissements et de fourmillements de l’extrémité distale d’un membre inférieur, puis apparaît un déficit des releveurs du pied et des orteils qui va entraîner rapidement un steppage à la marche.
Ce début est habituellement unilatéral, intéresse la loge antéroexterne de la jambe puis secondairement la loge postérieure, ce qui entraîne une diminution puis une abolition du réflexe achilléen.
L’atrophie musculaire et les fasciculations ne tardent pas à apparaître au niveau des muscles atteints.
Dans un délai variable, semaines ou mois, le membre controlatéral est atteint de la même manière, tandis que progresse l’amyotrophie sur le membre primitivement touché, gagnant le quadriceps.
Dans cette forme, le syndrome pyramidal est absent, seule une exagération des réflexes rotuliens pourrait dans certains cas déceler l’atteinte pyramidale, mais il existe des cas où ces réflexes sont faibles.
Devant un tel tableau, on conçoit très bien que le diagnostic de polynévrite soit posé.
La constatation de fasciculations est alors un appoint considérable pour le diagnostic.
L’évolution de cette forme est lente.
Ce n’est généralement qu’au bout de plusieurs mois que l’amyotrophie gagne les membres supérieurs, tandis que le syndrome pyramidal devient plus franc.
Ces formes n’ont pas reçu de la part des auteurs récents l’attention qu’elles méritaient.
Elle demeure même étrangement absente de la plupart des traités anglo-saxons.
Dans les études épidémiologiques, elle est généralement confondue avec le groupe des atrophies musculaires progressives ou les formes cliniques de SLA dont le début se situe aux membres inférieurs.
2- Atrophie musculaire progressive :
Elle est définie par l’atteinte progressive du neurone moteur inférieur sans altération clinique du neurone moteur supérieur.
Son existence est de plus en plus discutée en raison de nombreux cas d’atrophie musculaire progressive pour lesquels la vérification anatomique a pu montrer la présence d’une atteinte des voies corticospinales.
Le début est insidieux, l’atrophie musculaire débute le plus souvent par les membres supérieurs, de façon habituellement asymétrique (amyotrophie d’Aran-Duchenne).
Puis l’amyotrophie gagne les muscles du cou et les membres inférieurs.
Il n’y a pas de signe d’atteinte du système pyramidal et les réflexes tendineux sont précocement abolis.
Il peut exister au début des signes sensitifs.
Les formes à type d’atrophie musculaire progressive sont ainsi étudiées en commun avec les autres formes de maladie du motoneurone dans les études épidémiologiques récentes.
3- Forme bulbaire :
Il semble que, plus encore que pour l’atrophie musculaire progressive, les observations de lésions pures et isolées des noyaux bulbaires soient exceptionnelles.
De toute façon, l’évolution plus rapide de cette forme masque dans la plupart des cas les autres signes cliniques qui auraient pu éventuellement apparaître.
Les auteurs anglo-saxons, en accord avec la plupart des auteurs, l’intègrent dans le cadre des maladies du motoneurone, si ce n’est de la SLA.
Finalement, doit-on considérer que l’atrophie musculaire progressive et la paralysie bulbaire progressive sont des formes « extrêmes » et limitées d’une seule entité dont la SLA serait la forme la plus complète, la plus démonstrative et la plus fréquente ?
Cette opinion est partagée par la majorité des auteurs, en particulier anglo-saxons.
L’ignorance dans laquelle nous nous trouvons actuellement quant à l’étiologie, voire au mécanisme de ce type d’affection, doit nous rendre prudent dans l’établissement de classifications basées uniquement sur des critères cliniques.
4- Sclérose latérale primitive (ou forme pyramidale pure) : C’est une entité très discutée.
Elle a été initialement décrite par Charcot.
L’atteinte serait limitée aux faisceaux pyramidaux et aux motoneurones corticaux, principalement les cellules de Betz.
Certaines formes de maladies du neurone moteur à début pyramidal vont évoluer vers une SLA typique.
En revanche, dans des séries importantes, cette forme n’apparaît pas.
Des études récentes font état de lésions purement corticospinales sur la foi d’études cliniques et électromyographiques et anatomiques.
Dans les séries anatomiques, les cas ayant présenté une forme purement pyramidale avaient des lésions des motoneurones à la vérification anatomique.
Les critères de diagnostic ont été récemment proposés par Pringle et al.
Le début se fait à l’âge adulte, il est lentement progressif, sans à-coup, de façon symétrique, aux membres inférieurs ou dans la sphère bulbaire, caractérisé par une spasticité isolée, sans histoire familiale et sur une durée supérieure à 3 ans.
5- Formes monoméliques ou sclérose latérale amyotrophique bénigne ?
On connaît des formes de SLA dont l’évolution est très lente, 25 ans pour un malade de Charcot.
En dehors de ces aspects assez particuliers par la longueur de l’évolution, il existe des formes presque exclusivement amyotrophiantes, localisées à un segment de membre, encore appelées formes monoméliques par Rowland.
Elles sont caractérisées par une évolution lente ou encore par une stabilisation du processus, généralement limité à un membre supérieur.
Le début se fait habituellement dans l’enfance ou l’adolescence, la prédominance masculine est très nette (sans caractère familial).
La main et l’avant-bras sont touchés préférentiellement.
L’atrophie musculaire se développe progressivement sur 2 à 3 ans en l’absence de tout trouble sensitif.
Puis la maladie semble s’arrêter d’évoluer ou l’évolution devient très lente.
L’aggravation des troubles moteurs par l’exposition au froid a été signalée.
Dans certaines observations, un épisode fébrile précède le début des troubles et un tremblement accompagne l’atrophie.
Dans d’autres cas, plus rares, les membres inférieurs sont touchés et des douleurs et fasciculations ont été notées.
Les examens électriques dans tous ces cas confirmaient l’atteinte purement motoneuronale.
Aucun de ces cas n’a évolué vers un tableau de SLA, ce qui peut faire douter d’une quelconque relation entre les deux affections.
Il est cependant fort probable qu’il s’agisse d’un syndrome dont les étiologies sont diverses.
Certains cas sont familiaux, dont un qui relève d’une mutation de la SOD-1.
D’autres, sporadiques, pourraient être dus à une neuropathie motrice par bloc de conduction ou encore favorisés par une malformation du rachis cervical et des racines.
6- Autres modalités de présentation :
Les formes à début respiratoire méritent d’être individualisées.
L’atteinte des muscles respiratoires est classique et habituelle dans les stades ultimes de la maladie.
Elle réalise une hypoventilation alvéolaire.
Celle-ci est fréquemment décelable au moment du diagnostic, même dans les formes communes de SLA.
Dans la plupart des cas, une assistance respiratoire est souhaitable.
Elle entraîne rapidement une amélioration de la défaillance respiratoire.
Elle permet parfois une respiration indépendante au bout de plusieurs mois avec des épisodes de supports ventilatoires intermittents.
Dans certains cas, la détresse respiratoire est au premier plan ou isolée.
Le tableau est donc celui d’une insuffisance respiratoire aiguë souvent précédée de dyspnée et d’hypersomnie, le diagnostic de SLA étant alors le plus souvent méconnu.
Dans les cas où une vérification anatomique a été possible, on a constaté une atteinte des noyaux du nerf phrénique ou du noyau dorsal du vague à côté des lésions habituelles des cellules motrices de la corne antérieure.
Le déficit alvéolaire serait principalement dû à une faiblesse des muscles inspiratoires.
Parmi les autres types de présentation de la maladie, certains aspects apoplectiques ont été rapportés : début apparemment extrêmement brutal, en particulier bulbaire, faisant évoquer un accident ischémique.
La précession des signes habituels de la maladie par un amaigrissement a déjà été rapportée : Norris et al avaient en effet observé une perte de poids de plus de 50 % dans 5 % des cas de SLA et, chez deux patients, une perte de plus de 30 % était le symptôme initial de la maladie.
7- Aux limites des formes cliniques de la maladie :
La présence des signes classiquement absents peut orienter le diagnostic vers d’autres affections.
Dans les atteintes du tronc cérébral, on a vu la prédominance ou l’exclusivité des lésions dans la région bulbaire, mais d’autres structures peuvent être exceptionnellement touchées.
* Troubles oculomoteurs :
Les noyaux oculomoteurs sont indemnes dans la presque totalité des cas.
Les vérifications anatomiques de telles observations sont exceptionnelles. Lawyer et Netsky rapportèrent des lésions des noyaux oculomoteurs dans quatre de leurs 53 cas de SLA (sans anomalies oculomotrices cliniques).
C’est Harvey et al, qui, dans un cas de SLA ayant présenté des troubles oculomoteurs précoces dans l’évolution de la maladie, montrèrent l’existence de lésions dans les noyaux des IIIe, IVe et VIe nerfs crâniens.
Des études récentes ont montré dans des cas de SLA un trouble des mouvements oculaires sous forme d’un déficit des mouvements oculaires de poursuite étudiés par électro-oculographie.
Dans 11 des 18 cas étudiés par Jacobs et al existait un tel trouble ; il existait en plus chez trois d’entre eux une perturbation des saccades.
Dans un de ces cas avec trouble de poursuite, l’autopsie révéla l’existence de lésions de la substantia nigra et de la capsule interne.
Leveille et al trouvèrent également des troubles des saccades ou de poursuite dans quatre cas sur dix.
Ces auteurs suggèrent que ces troubles sont d’origine supranucléaire et que le système oculomoteur est plus souvent atteint dans la SLA que précédemment reconnu.
Dans 15 cas sur 24 patients atteints de SLA, Esteban et al trouvèrent une altération du phénomène de Bell.
Trois cas avaient en plus une altération de la motilité conjuguée oculaire vers le haut, l’ensemble de ces troubles évoquant des lésions corticonucléaires.
Kondo et Hemmi notent la présence de troubles des mouvements oculaires, sans autres précisions, dans 3,6 % des cas.
L’existence d’un nystagmus n’a, en revanche, été rapportée qu’exceptionnellement (2,6 %des cas pour Kondo et Hemmi). Kushner et al en rapportent deux cas avec vérifications anatomiques. Dans l’un de ces cas, une parésie du regard était également présente.
Dans des cas de manifestation complète de la maladie, des troubles oculomoteurs surviennent, ils sont dans ces conditions dus à des lésions supranucléaires.
* Troubles sphinctériens :
Ils sont très rares, même aux stades ultimes de la maladie.
Tsubaki les note dans 1 % des cas, de même que Jokelainen.
Gubbay et al et Kondo et Hemmi les notent respectivement dans 4 et 5 % des cas.
L’absence ou la discrétion des troubles sphinctériens dans la SLA serait due à l’intégrité des noyaux sacrés d’Onuf.
* Troubles sensitifs :
Dans la SLA, ils ne sont pas exceptionnels.
Charcot avait déjà souligné leur présence, tout du moins aux stades initiaux de la maladie, sous forme d’engourdissements ou de fourmillements.
L’existence d’une démyélinisation des cordons postérieurs a été abondamment rapportée dans des observations authentiques de SLA en l’absence de tout signe sensitif objectif ou subjectif.
De même, des altérations des fibres sensitives ont été observées à l’occasion de biopsies nerveuses.
Dans les grandes séries de SLA rapportées, les troubles sensitifs ont été observés avec une fréquence variable : 2,8 % pour Gubbay et al ; 4 % pour Tsubaki ; 7,4 % pour Kondo et Hemmi.
Des études utilisant des seuils thermiques par microprocesseurs automatisés ont montré l’existence d’anomalies des sensibilités thermiques dans 80 %des cas, indiquant ainsi une atteinte des fibres sensitives de petit calibre.
Les fibres de plus gros calibre sont également touchées, comme le montrent la présence de démyélinisation dans les cordons postérieurs et l’existence d’altérations de la conduction lors de l’étude des potentiels évoqués somesthésiques.
Des douleurs sont parfois rencontrées, essentiellement lors de la phase terminale de la maladie, où, selon Newrick et Langton-Hewer, 40 % des patients atteints de SLA se plaindraient d’hyperesthésie, de névralgie, de douleur articulaire, de crampes et de pseudopolyarthrite de l’épaule.
* Troubles vasomoteurs : Ils sont assez fréquents.
Ils siègent aux extrémités des membres supérieurs et inférieurs.
Celles-ci sont froides, cyanosées, parfois oedématiées.
Une atteinte objective du système nerveux autonome est rarement observée, peut-être par manque d’études systématiques.
En effet, des études récentes ont montré des altérations de certaines fonctions du système nerveux autonome.
Ces anomalies ont été retrouvées dans 12 % des cas par Tsubaki.
Faut-il rattacher l’absence d’escarres, même aux stades ultimes de la maladie, à la discrétion ou à l’absence de troubles sensitifs et végétatifs ?
Toujours est-il que cette constatation est habituelle et reconnue par de nombreux auteurs. Sur 36 patients atteints de SLA, hospitalisés et confinés au lit, aucun ne développa d’escarres.
Furukawa et Toyokura suggèrent, après étude pléthysmographique des doigts, qu’il existe une constriction artériolaire chez les patients atteints de SLA, ce qui serait peu propice au développement d’escarres.
* Troubles psychiques :
Rencontrés dans la SLA, ils sont actuellement reconnus comme fréquents et de plus en plus étudiés.
Si l’on exclut les cas de démence qui seront étudiés plus loin, les troubles psychiques sont relativement peu marqués.
Gubbay et al notaient chez six patients une labilité émotionnelle, tandis que 20 patients de la même série (sur 318) avaient des troubles d’allure démentielle.
Jokelainen, sur 39 patients (15 % des cas de sa série) présentant des altérations psychiques, observait : une labilité émotionnelle dans 36 % des cas, un syndrome dépressif dans 26 %, un comportement euphorique dans 15 %, un état confusionnel dans 8 %, un état paranoïaque dans 3 %et un état véritablement démentiel dans 13 % des cas.
Des manifestations d’allure frontale ont été récemment mises en évidence par des tests psychométriques seuls, soit associés à des études par imagerie : résonance magnétique (IRM) et positon emission tomography (PET) scanner.
F – Scléroses latérales amyotrophiques familiales :
La fréquence des formes familiales varie selon les séries ; elle représenterait environ 5 à 10 % des cas de SLA.
Hudson étudia les cas rapportés de 1955 à 1980 : 61 familles représentant 270 cas, parmi lesquelles trois familles étaient à début juvénile et dix comportaient une démence et/ou un syndrome parkinsonien.
Si l’on examine les caractères épidémiologiques des 210 cas restants (48 familles), on observe que le rapport H/F est de 1,3/1, l’âge moyen de début de 47,3 ans et la durée moyenne d’évolution de 4,1 années.
Horton et al, après analyse de 14 familles venant de leur propre recrutement, distinguèrent au moins trois types différents : le premier correspond aux formes classiques de SLA à évolution rapide ; le deuxième type comprend des patients dont l’évolution est également courte mais qui présentent en plus une atteinte des cordons postérieurs, voire spinocérébelleux avec parfois des inclusions hyalines, le troisième type est représenté par une évolution beaucoup plus lente mais avec également une atteinte des cordons postérieurs.
Dans une étude portant sur 103 patients avec SLA familiale observés à la Mayo Clinic, les auteurs notaient qu’il n’existait pas de différence significative entre les formes sporadiques et familiales quant à l’évolution clinique et qu’il n’y avait que des différences mineures quant à l’âge de début, le rapport H/F, la durée de survie et l’atteinte initiale aux membres inférieurs.
En reprenant les cas familiaux publiés dans la littérature, soit 206 familles représentant près de 1 000 sujets atteints, Moulard et al ont tracé les caractères cliniques évolutifs principaux de la forme familiale de SLA comparée à la forme sporadique.
Dans la plupart des familles, la transmission apparaît comme autosomique dominante.
Dans de grandes familles de SLA, une liaison sur le chromosome 21 (q21.1- q22.3) a été mise en évidence.
Rosen et al ont identifié les mutations du gène responsable.
Il s’agit du gène SOD-1 codant pour l’enzyme SOD.
La fonction de la SOD est de convertir les anions radicaux libres superoxydes en hydrogène peroxyde.
La forme de SOD impliquée dans les formes familiales de SLA est l’enzyme cuivre/zinc contenue dans les cytosomes.
Il existe plus de 40 mutations du gène SOD-1 aujourd’hui rapportées. Une mutation du gène SOD-1 est présente dans 15 à 20 %des formes familiales de SLA.
Il semble exister quelques variations phénotypiques en rapport avec certaines mutations ; notamment dans les familles japonaises avec la mutation His46Arg donnant lieu à des formes de survie prolongée ou encore la mutation Gly93Ser donnant des symptômes sensitifs et végétatifs prédominants.
Il semble exister une grande hétérogénéité phénotypique dans les formes familiales de SLAavec mutations du gène SOD-1.
G – Sclérose latérale amyotrophique et démence :
La fréquence de cette association est de l’ordre de 5 % ou moins.
Elle est de 4 % pour Tsubaki et de 6,2 % pour Gubbay et al.
Dans la revue de cas antérieurement publiés, Hudson a relevé cette association au cours de SLA sporadiques dans 42 cas se répartissant ainsi : 26 cas avec démence et SLA, huit avec syndrome parkinsonien et SLA, huit avec démence, syndrome parkinsonien et SLA.
Dans la majorité des cas, le tableau démentiel est celui d’une démence présénile de type frontal, plus rarement de type Alzheimer ou Pick.
Le rapport H/F est de 2,2/1 ; l’âge moyen de début de la SLA est de 53,3 ans ; l’âge moyen de début de la démence est de 52,1 ; la durée moyenne d’évolution de la maladie est de 2,1 années.
Le tableau clinique de la SLA n’offre rien de particulier et est superposable à celui de la forme commune. Brion et al, à propos d’un cas personnel, ont repris tous les cas antérieurement publiés de maladie de Pick (12 cas) associés à une SLA.
Ils proposent trois mécanismes : une simple coïncidence des deux processus, l’extension du processus atrophique de la maladie de Pick à la frontale ascendante ou encore un début atypique hors de la frontale ascendante d’un processus de SLA.
Dans les cas observés au Japon et rapportés par Mitsuyama, la démence ne présentait aucun caractère spécifique, ni clinique, ni anatomique et l’aspect anatomopathologique de la SLAn’offrait aucune différence significative avec les aspects habituellement observés dans les formes sporadiques communes de SLA.
Les tests pyschométriques ont montré que les troubles de la mémoire relevaient plutôt de difficultés d’attention, sans doute de type frontal.
Afin de résoudre le problème d’une éventuelle association entre SLA et maladie de Creutzfeldt-Jakob, Salazar et al ont revu plus de 2 000 cas de cette maladie et des troubles associés rapportés dans la littérature et 231 cas de démence associés à une atteinte motoneuronale précoce provenant de leur recrutement personnel.
Dans la plupart des cas, la démence associée est le plus souvent liée à la forme classique de SLA et aucun ne semblait pouvoir correspondre à la maladie de Creutzfeldt-Jakob transmissible.
Lorsqu’une atteinte motoneuronale apparaît dans l’évolution d’une maladie de Creutzfeldt-Jakob, elle est souvent tardive et accompagnée de signes d’atteintes cérébrale et cérébelleuse fulminantes.
H – Formes rencontrées dans le Pacifique Ouest :
La SLA a une incidence et un taux de mortalité d’approximativement 1 pour 100 000 habitants.
Cette incidence, à l’exception de quelques rares régions, est à peu près uniformément retrouvée dans tous les pays.
Les exceptions sont situées dans des îles du Pacifique Ouest : l’île de Guam, la péninsule Kii au Japon et les régions ouest de la Nouvelle-Guinée où l’incidence est quelque 100 fois celle de la forme sporadique.
L’île de Guam est la plus grande des îles de l’archipel des Mariannes, qui, avec entre autres les îles Carolines, font partie de la Micronésie.
Les natifs de l’île de Guam appartiennent au groupe linguistique Chamorro et descendent probablement de peuplades aborigènes d’origine malaisienne ou indonésienne.
Les Chamorros connaissaient la SLA qu’ils appelaient « lytico », elle-même connue dès le début du XIXe siècle.
L’existence d’une paralysie héréditaire dans l’île de Guam fut rapportée pour la première fois en 1900. Dès 1931, cette affection était appelée SLA sur les certificats de décès. Les études modernes datent de 1945.
En 1956, un centre permanent de recherche était établi dans l’île.
L’association maladie de Parkinson-démence, nommée bodig, a été pour la première fois observée en 1954 et reconnue comme une nouvelle maladie en 1959.
De nombreuses revues ont par la suite été publiées, tant sur les aspects cliniques qu’épidémiologiques et tant pour la SLAque pour le syndrome de Parkinsondémence .
En dehors de l’île de Guam, deux autres foyers à haute incidence de SLA et de Parkinson-démence ont été découverts : l’un au Japon, dans les villages Hobara et Kozagawa de la péninsule Kii de l’île de Honshu ; l’autre dans les plaines côtières du sud de la région ouest de la Nouvelle-Guinée dans les groupes culturels et linguistiques auyu et jakai.
Trois autres foyers à haute incidence de maladies neurologiques dégénératives souvent illustrées par une amyotrophie ont été récemment découverts : chez les aborigènes d’Australie à Groote Eylandt et la côte adjacente de la terre d’Arnehm dans le golfe de Carpentaria ; dans la population iakoute de la vallée de la rivière Viliui en Iakoutie (URSS) et chez les Philippins en provenance des provinces Iliocos et immigrant à Hawaï.
Les aspects cliniques et évolutifs de SLA survenant dans l’île de Guam, au Japon dans la péninsule Kii et en Nouvelle-Guinée diffèrent peu de la forme classique sporadique. Dans quelques cas (cinq sur 104), la SLA s’enrichit en cours d’évolution de signes extrapyramidaux et démentiels.
À l’inverse, sur 72 cas de syndrome Parkinson-démence, 27 se sont enrichis en cours d’évolution (en moyenne 1,7 an après le début) de signes de SLA.
Dans l’étude comparative réalisée par Hirano et al entre les cas de l’île de Guam et les SLA sporadiques observées à New York, l’aspect clinique de la maladie était sensiblement identique, mais la durée d’évolution était différente chez les Chamorros : une durée d’évolution de plus de 5 ans était retrouvée dans plus d’un tiers des cas.
L’existence de tels foyers a suscité de nombreuses recherches afin de découvrir l’éventuel dénominateur commun à ces maladies et la cause de la SLA.
En effet, l’existence de ces isolats géographiques à très haute incidence laisse supposer qu’un facteur local environnemental (toxique, viral, nutritionnel) est à l’origine de cette affection.
L’hypothèse génétique a été soulevée à cause de l’agrégation familiale.
Mais aucun argument décisif n’a jusqu’à présent été apporté, si bien que les recherches se sont orientées vers la mise en évidence d’un facteur environnemental.
La recherche d’un agent viral a pourtant toujours été négative.
Toutes les tentatives faites pour isoler un virus ou pour transmettre la maladie à l’animal ont échoué.
Il en est de même de la recherche d’un déficit immunologique, quoique, si l’immunité cellulaire s’est avérée être normale, il a été trouvé des anomalies de l’immunité humorale, en l’occurrence une augmentation sérique des immunoglobulines (Ig) A et IgG sans rapport avec une réponse spécifique antivirale ou auto-immune mais plutôt en rapport avec des infections répétées et une immunodéficience, chez les patients atteints de SLA.
Une origine toxique, en particulier due à l’ingestion de cycade dont certains composants (méthylazoxyméthanol) seraient toxiques pour le système nerveux, a été retenue par certains, mais aucune relation entre SLA et cycade n’a été sérieusement démontrée.
Parallèlement à ces recherches, les mouvements migratoires vers et en provenance de l’île de Guam ont été particulièrement étudiés.
Garruto et al observèrent un tableau de SLA chez 28 Chamorros migrants dont 24 ont vu leur maladie débuter aux États-Unis, au Japon, en Allemagne et en Corée après des périodes d’absence de 1 à 34 ans de l’île de Guam.
La période de latence de la maladie, si celle-ci est due à des facteurs environnementaux dans l’île de Guam, serait d’une trentaine d’années.
Quatre autres patients ont développé la maladie dans les 1 à 14 ans suivant leur retour à Guam, après un long séjour aux États-Unis.
Le temps d’exposition minimal aux facteurs environnementaux dans l’île de Guam serait de 18 ans.
Cette incidence parmi les populations migrantes de Chamorros a également été reconnue par Eldridge et al ; pour ces auteurs, ces constatations seraient compatibles avec l’hypothèse d’une origine génétique ou d’une exposition précoce à des toxiques environnementaux dans l’île de Guam ou encore les deux à la fois.
Garruto et al notèrent ainsi une augmentation du nombre de cas de SLA chez les Philippins ayant émigré à Guam depuis de longues années.
Mais près de la moitié de ces immigrants venaient de la province Iliocos dont on sait que de nombreux Philippins qui en sont issus, migrant à Hawaï, ont développé cette affection, ce qui laisserait supposer une prédisposition génétique des populations « Iliocos » ; mais comme la prévalence de la SLA dans ces régions est inconnue, l’hypothèse d’un facteur environnemental à Guam demeure toujours prédominante.
Un tel déclin est également observé dans la péninsule Kii au Japon, bien que l’incidence reste élevée et l’âge de début plus tardif.
L’explication de ce phénomène est proposée par Garruto et al : il serait dû aux nouvelles habitudes de vie et alimentaires de ces populations en raison de l’occidentalisation accélérée de leur mode de vie.
Des articles plus récents font cependant état de résultats moins catégoriques.
Les deux syndromes, lytico et bodig, demeurent prévalents dans l’île de Guam.
Les âges de début et de décès sont plus tardifs, sinon les autres caractéristiques de la maladie n’ont pas changé.
Dans un précédent article, Yanagihara et al avaient déjà soulevé l’hypothèse que la faible teneur en calcium et en magnésium de l’eau et du sol rencontrée dans ces régions (mais aussi dans la péninsule de Kii et en Nouvelle-Guinée), entraînait un hyperparathyroïdisme secondaire et un déficit minéral dû au déficit nutritionnel chronique en calcium et magnésium.
Cela conduirait à une absorption intestinale accrue de calcium et d’autres métaux et entraînerait ainsi une déposition de calcium et d’aluminium dans le tissu nerveux. Perl et al ont montré une accumulation intraneuronale d’aluminium dans la SLA et le syndrome Parkinson-démence des Chamorros.
Actuellement, les recherches sont particulièrement focalisées sur les cas existant en Nouvelle-Guinée où l’incidence demeure très élevée, dix fois celle de Kii et de Guam, et sur les cas découverts dans d’autres foyers à haute incidence de SLA ou de maladies voisines.
C’est le cas chez les aborigènes d’Australie de Groote Eylandt où coexistent deux affections distinctes mais peut-être liées : l’une se présente par un grand syndrome cérébelleux, un syndrome pyramidal et parfois une paralysie oculaire supranucléaire et une démence ; l’autre débute dans l’enfance, elle est caractérisée par une laxité ligamentaire marquée et une atteinte pyramidale et des motoneurones.
La survenue de ces deux affections dans la même famille les fait rapprocher du même syndrome observé à Guam.
En Iakoutie, bien que les informations soient rares, il ressort que la maladie est connue depuis une centaine d’années et affecte 1 % de la population des villages touchés.
Le début est aigu, fébrile avec léthargie.
C’est un tableau d’encéphalite avec ophtalmoplégie, syndrome extrapyramidal, ataxie et troubles psychiques.
Le liquide céphalorachidien (LCR) montre une hypercytose faite de lymphocytes (1 à 50).
Cette phase dure de quelques jours à quelques mois.
Elle est suivie, après une période de récupération totale ou partielle de plusieurs semaines ou mois, par un syndrome fatal en 2 à 5 ans, marqué par une démence, une spasticité, une rigidité extrapyramidale et des signes d’atteinte des nerfs crâniens.
Des signes cérébelleux et surtout une amyotrophie sont rencontrés dans les formes chroniques.
À l’autopsie, il existe une hydrocéphalie et une atrophie corticale, une gliose diffuse, des signes inflammatoires, des plaques amyloïdes et des aspects de dégénérescence neurofibrillaires.
Tous ces éléments cliniques et anatomiques militent en faveur d’une étiologie infectieuse (virale ?) ; mais la preuve n’en a pas été apportée à ce jour.
Ainsi, les études faites sur ces isolats à haute incidence laissent espérer pour l’avenir un éclaircissement des possibles facteurs environnementaux et génétiques responsables de ces différents syndromes dont il n’est cependant pas certain qu’ils puissent être de même nature que la SLA sporadique rencontrée dans les autres régions.
I – Formes juvéniles de sclérose latérale amyotrophique :
La plupart des formes juvéniles rapportées sont familiales.
Les formes sporadiques diffèrent peu de la forme classique de l’adulte.
Dans une revue de cinq cas rapportés dans la littérature, Nelson et Prensky ne notèrent pas de différences cliniques significatives ; la présence d’inclusions neuronales contenant de l’acide ribonucléique (ARN) laissait supposer à ces auteurs qu’il s’agissait peut-être d’une forme nosologiquement distincte de la forme de l’adulte.
Dans les formes familiales recensées par Emery et Holloway, le mode de transmission était autosomique récessif dans cinq familles et dominant dans le reste (cinq familles).
La durée d’évolution semble être beaucoup plus longue que dans la forme adulte.
Le début des troubles se fait dans l’enfance ou l’adolescence. Les formes récessives ont un début plus précoce (3,3 ± 2 ans) que les formes dominantes (11,5 ± 8,4 ans).
L’atteinte bulbaire est présente dans 80 % des cas récessifs et très rare dans les cas dominants.
Dans les formes tunisiennes, la transmission apparaît comme autosomique récessive avec une moyenne d’âge de début de 12 ans, et au moins trois formes de présentation ont été décrites : une avec amyotrophie des membres supérieurs et atteinte pyramidale et bulbaire, une autre avec seulement l’atteinte bulbaire et enfin une troisième avec atrophie péronière et spasticité des membres inférieurs.
Une liaison génétique au chromosome 2q33-q35 a été trouvée dans certaines de ces formes récessives tunisiennes.
Dans une forme autosomique dominante, une liaison au chromosome 9q34 a été observée.
Dans le sud de l’Inde, une forme juvénile sporadique particulière a aussi été décrite.
Elle associe une atteinte de type périphérique et bulbaire à une surdité.
L’évolution est lentement progressive et relativement bénigne.
J – Amyotrophies postpoliomyélite :
Certains patients atteints de poliomyélite semblent être deux fois « maudits » ; non seulement ils souffrent d’une amyotrophie due à la maladie virale initiale, mais encore d’une atrophie musculaire progressive insidieuse qui peut se développer plusieurs années plus tard.
Campbell et al rapportent cinq cas de dégénérescence motoneuronale faisant suite à une ancienne poliomyélite.
À cette occasion, ils font une revue de 83 cas antérieurement publiés dans la littérature.
La maladie présente une évolution relativement bénigne, tous les patients étant vivants au moment des publications, avec un recul moyen de 4,6 années.
Hayward et Seaton ont étudié 24 volontaires ayant présenté une poliomyélite jusqu’à 51 ans auparavant.
L’étude électrophysiologique quantitative a montré des anomalies de dénervation chronique dans des muscles apparemment sains.
Il est suggéré qu’il existe dans ces cas une accélération de la déperdition neuronale avec l’âge.
Wiechers et Hubbell ont étudié en fibre unique dix patients ayant présenté une poliomyélite auparavant mais ne souffrant d’aucune faiblesse nouvelle.
Les anomalies découvertes pourraient témoigner d’une désintégration avec l’âge des unités motrices réinnervées.
Enfin, dans un cas de SLA survenue 32 ans après une attaque de poliomyélite, Roos et al ont trouvé à l’autopsie des lésions typiques de SLA sans inclusion ni signe inflammatoire.
Il pourrait ainsi exister un syndrome postpoliomyélitique, rare, dont l’étiologie est inconnue, mais qui, pour certains serait dû à une défaillance des capacités de réinnervation.
Les essais entrepris pour isoler ou transmettre le virus ont été négatifs.
Finalement, le rôle du virus de la poliomyélite dans l’étiologie des atrophies musculaires progressives postpoliomyélitiques et dans la SLA demeure entièrement hypothétique.
Des études récentes n’ont en effet pas confirmé la plus grande fréquence de SLA chez des patients préalablement atteints de poliomyélite.
D’autre part, la poliomyélite pourrait avoir au contraire un rôle protecteur contre le développement d’une SLA.
Il ne semble pas, en outre, y avoir de progression significative du déficit neurologique chez les survivants d’anciennes paralysies poliomyélitiques.
K – Sclérose latérale amyotrophique post-traumatique :
Le rôle des traumatismes a déjà été envisagé comme facteur de risque dans le chapitre Épidémiologie.
Quant à la réalité de SLA post-traumatiques, la prudence est de mise et aucune preuve n’a à ce jour pu être apportée de cette association.
L – Sclérose latérale amyotrophique et néoplasmes (SLA paranéoplasiques ?) :
Norris et Engel en 1965, dans une étude sur 130 cas de SLA, trouvèrent 13 fois un néoplasme associé, soit dix fois plus que dans une série comparable de patients atteints de maladies vasculaires cérébrales.
Dans trois cas, il s’agissait de cancers du poumon, dans deux de cancer du côlon et un cas de séminome, thymome, lymphosarcome, astrocytome, adénocarcinome de la prostate, leucémie, carcinome basocellulaire et adénocarcinome du sein.
Il y avait 11 hommes pour deux femmes. Huit d’entre eux furent autopsiés et dans aucun cas un envahissement direct par le processus néoplasique ne pouvait rendre compte du tableau de SLA.
Brain et al, la même année, rapportaient 11 cas de SLA associée à un néoplasme dont cinq cancers du poumon et trois du sein.
Deux cas ont été autopsiés et les résultats différaient des formes typiques de SLA ; les lésions dégénératives étaient moins marquées et il existait des modifications notables des cordons postérieurs.
Buchanan et Malamud ont rapporté, en 1973, l’observation d’un jeune homme de 36 ans qui a présenté, 2 ans et demi avant la découverte et l’ablation d’un cancer du rein, un déficit et une atrophie du membre inférieur droit qui rapidement se sont étendus, avec des fasciculations diffuses.
Le tableau neurologique s’est aggravé après l’intervention puis est demeuré stable jusqu’au décès du patient dû à des métastases, plusieurs années plus tard.
À l’autopsie, il existait des modifications dégénératives chroniques des motoneurones sans réaction inflammatoire.
Mitchell et Olczak rapportaient également, en 1979, l’observation d’un homme de 54 ans qui présentait une amyotrophie progressive des membres inférieurs depuis 6 mois avant la découverte d’un cancer du poumon.
À l’examen, il existait des fasciculations, une atrophie musculaire étendue et des réflexes très vifs. Le LCR était normal et l’examen électromyographique était en faveur d’une affection du motoneurone.
Après l’intervention chirurgicale, les fasciculations disparurent et 6 mois plus tard, le tableau clinique s’était nettement amélioré. L’examen électrique pratiqué alors pouvait être considéré comme normal.
Dans l’étude rétrospective de la littérature faite par Barron et Rodichok, l’incidence de l’association SLA-cancer varie grandement selon les séries.
Les symptômes initiaux étaient bulbaires dans 30 % des cas, aux bras dans 35 % et aux jambes dans 35 %. Le néoplasme était contemporain dans deux cas et précédait la SLA dans trois cas.
Dans une étude ultérieure portant sur un plus grand nombre de cas, Norris retrouvait une incidence de 6 % de cancer pour 300 patients avec SLA.
Finalement, à la lecture de ces résultats et à partir d’études plus récentes, il apparaît que l’incidence du cancer chez les patients atteints de SLA n’est pas plus grande que ce que l’on attend chez des témoins du même âge.
Quelques cas particuliers sont cependant troublants d’association cancer-maladie du motoneurone, d’autres sontmêmeconvaincants dans la mesure où l’atteinte motoneuronale disparaît après traitement du néoplasme.
Finalement, Forsyth et al concluent que si l’association cancer-maladie du motoneurone est dans la majorité des cas fortuite, des cas particuliers d’association, notamment s’il existe des anticorps anti-Hu, sont possibles.
Dans les lymphomes, Walton et al avaient déjà rapporté l’observation d’une affection en tout point semblable à une atrophie musculaire progressive subaiguë associée à une maladie de Hodgkin.
L’autopsie révélait l’existence de lésions évocatrices d’une affection virale et des particules virus-like furent identifiées dans les cornes antérieures de la moelle.
Par la suite, Schold et al publièrent les observations de dix cas assez semblables mais qui différaient cependant par une évolution relativement bénigne et l’atteinte motoneuronale subaiguë semblait être indépendante de l’affection hématologique.
L’hypothèse virale était retenue pour la plupart des cas.
Des publications plus récentes font état d’association entre SLAet lymphome, le plus souvent associés à une paraprotéinémie.
Dans tous les cas existait une élévation de la protéinorachie.
À ce jour, plus d’une cinquantaine de cas de lymphomes ou de syndromes lymphoprolifératifs ont été rapportés.
Dans la majorité des cas, le traitement du lymphome n’a pas entraîné d’amélioration de l’état neurologique.
Pour les gammapathies monoclonales bénignes ou malignes, le tableau clinique est plus proche de la SLA classique. Bauer et al rapportent le cas d’une SLA au cours d’une maladie de Waldenström avec, à l’autopsie, des lésions dégénératives typiques telles qu’on les rencontre dans les SLA sporadiques
Le cas de Rowland et al est assez semblable avec des lésions anatomiques de la corne antérieure typiques de SLA.
Dans le cas de Krieger et Melmed, il s’agissait d’une gammapathie monoclonale à IgG, les lésions anatomiques étaient identiques aux cas précédents.
Patten rapporte trois cas de SLA associées à une gammapathie à IgG : dans deux d’entre eux, le syndrome clinique de SLA a régressé sensiblement après traitement de la gammapathie, entraînant une réduction du taux de la gammaglobuline sérique.
Cependant, les associations SLA et gammapathie monoclonale IgG semblent n’être qu’une coïncidence.
Une élévation des anticorps polyclonaux IgM antigangliosides a été, a plusieurs reprises, mise en évidence chez des patients avec SLA.
Les amyotrophies postradiothérapie, dont certains cas ont pu être attribués à une atteinte du motoneurone dans la moelle, semblent en fait être dues à des lésions des racines nerveuses motrices.





